Welcome to the Shin Lab research page at the University of Toronto!
We take a problem-solving approach to our research. We study how proteins, a natural scaffold for molecular recognition, interact with specific ligands when there are so many to choose from in the cell. In order to gain an understanding of this large problem, we use a diverse array of tools from molecular & cellular biology, biochemistry & biophysics, organic, analytical, & physical chemistry. We apply our knowledge to protein design, in particular, design of proteins that target specific DNA sites. We test their ability to function by in vitro assays (e.g. gels, blots, spectroscopy) and in vivo assays (e.g. bacteria, yeast, mammalian cells, and mouse models). With chemical and molecular biological tools, we are designing new proteins capable of selectively binding desired DNA sequences. These designed proteins may eventually lead to new drugs that target specific diseases with fewer side effects.
Some examples of experiments and techniques are shown below. These techniques allow us to test our hypotheses about structure-function relationships in biomolecular assemblies. Click on the photos to see enlarged data graphics.
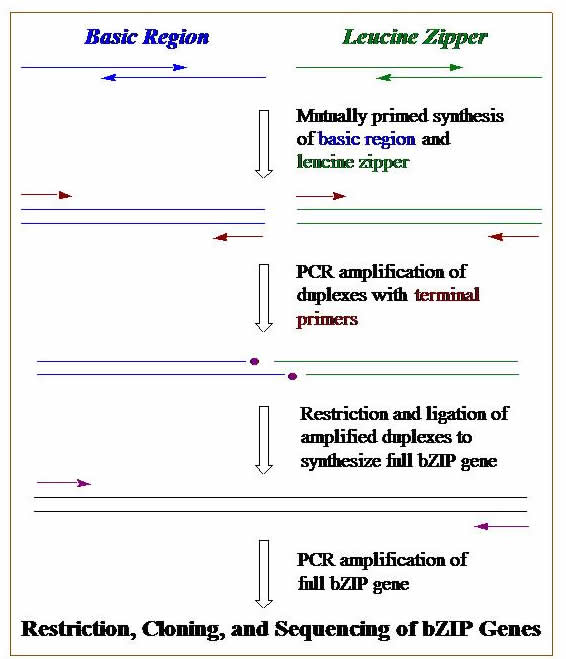
DNA Synthesis, Cloning, & PCR. We construct genes that code for our designed proteins. These genes can be synthesized on a DNA synthesizer, by using polymerase chain reaction (PCR), or a combination of both. Genes are then cloned into plasmid DNA. Bacterial cells are transformed with these engineered plasmids, and these cells become mini factories for our proteins. The graphic outlines the basic steps we use to construct a gene using DNA synthesis, PCR, cloning, & transformation.

Protein Expression & Purification. After growing protein in transformed cells, the cells are lysed and thecontents purified. This can be seen in the SDS-PAGE gel showing the purification of protein through different steps: molecular weight markers (Lane 1), crude cell lysate (Lane 2), after purification on a metal-ion affinity column (Lane 3), and after size-exclusion chromatography (Lane 4). After the final purification shown in Lane 4, note the purity of protein (denoted by the red arrow).

Chromatography: HPLC, FPLC, ion-exchange, size-exclusion. Typically, more than one type of chromatography is needed to purify our proteins. After metal-ion affinity chromatography, we commonly use reverse-phase HPLC or size-exclusion chromatography, which can be seen in the graphic. Impurities come out in the low, broad peak before the peak we collect, which is marked with the horizontal line.
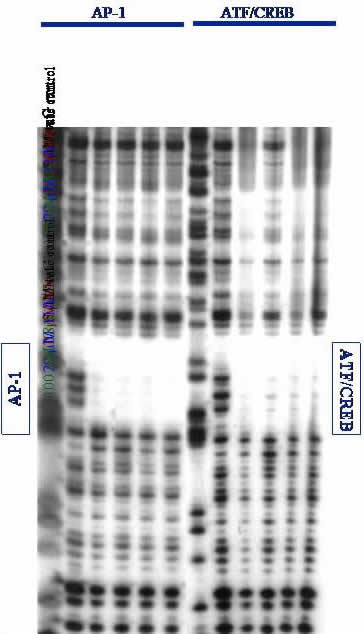
Gel Electrophoresis. DNA is separated on low-resolution agarose gels or high-resolution polyacrylamide gels (PAGE). Proteins are visualized on PAGE gels followed by immunoblot assay. A high-resolution DNase I footprinting gel is shown; if our protein (or ligand) binds to a specific DNA site, it will protect that site from random DNase I cleavage. This can be seen by the empty space or "footprint" left by our proteins binding specifically to the AP-1 and ATF/CREB sites on a ~650 base-pair fragment of DNA. Footprinting is an especially stringent technique for evaluating specific binding of ligands to DNA. From this data, we conclude that our proteins can specifically bind to desired DNA sites.
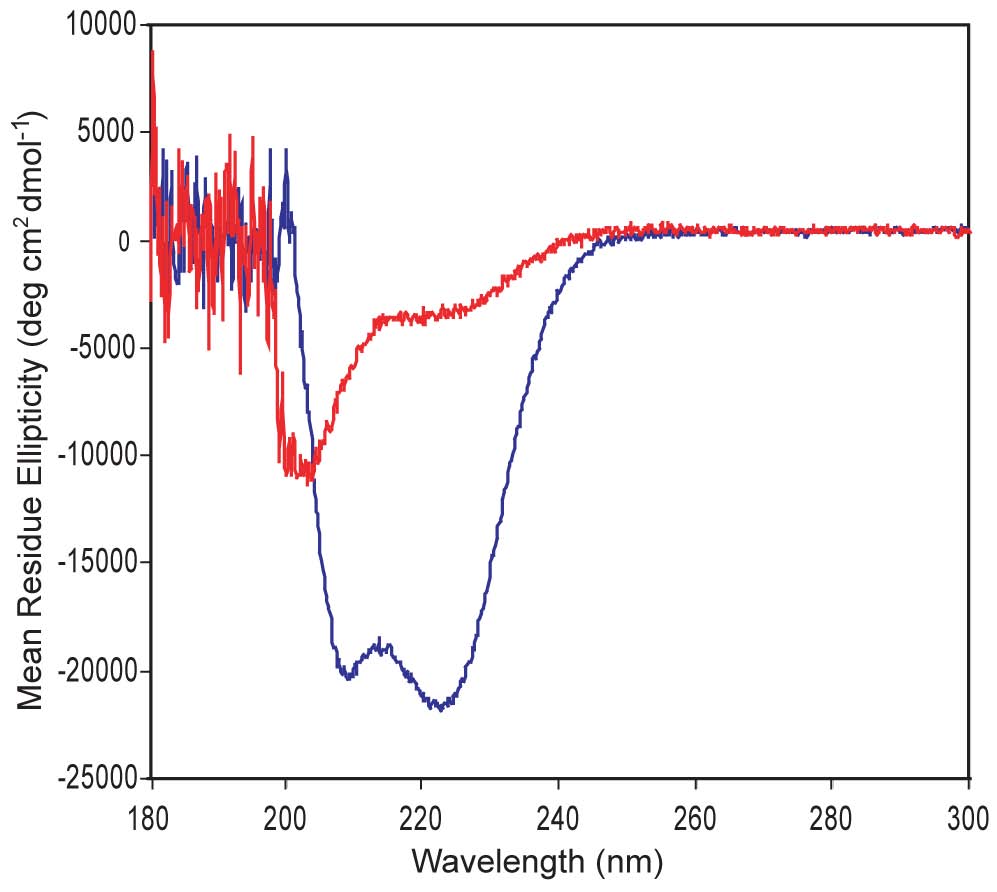
Circular Dichroism. We are designing a-helical proteins. CD is an ideal technique to evaluate a-helices, which display double minima at 208 nm & 220 nm. The CD shown here confirms that one of our proteins shows good a-helical structure (blue) while the other is poorly structured (red). A major question we ask is whether good a-helical structure is necessary for strong DNA-binding function and whether a protein's helical structure increases upon DNA binding.
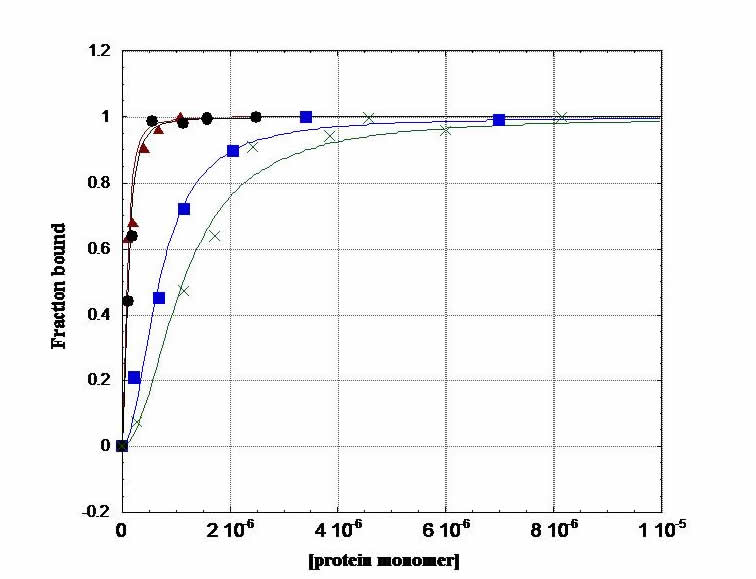
Fluorescence Anisotropy. In an anisotropy titration, we add aliquots of protein to fluorescein-labeled DNA in a cuvette. As protein concentration increases, binding to DNA increases and increases the anisotropy of the fluoresceinated DNA: i.e. as the protein binds, the tumbling motion of DNA in solution slows down. This experiment gives us information on the thermodynamics of protein-DNA complexation. We generate a binding isotherm that provides us with the free energy of binding in our system. This is a terrific experiment; you can do a titration and fit the data on the computer, and within a couple of hours, you have your free energy.
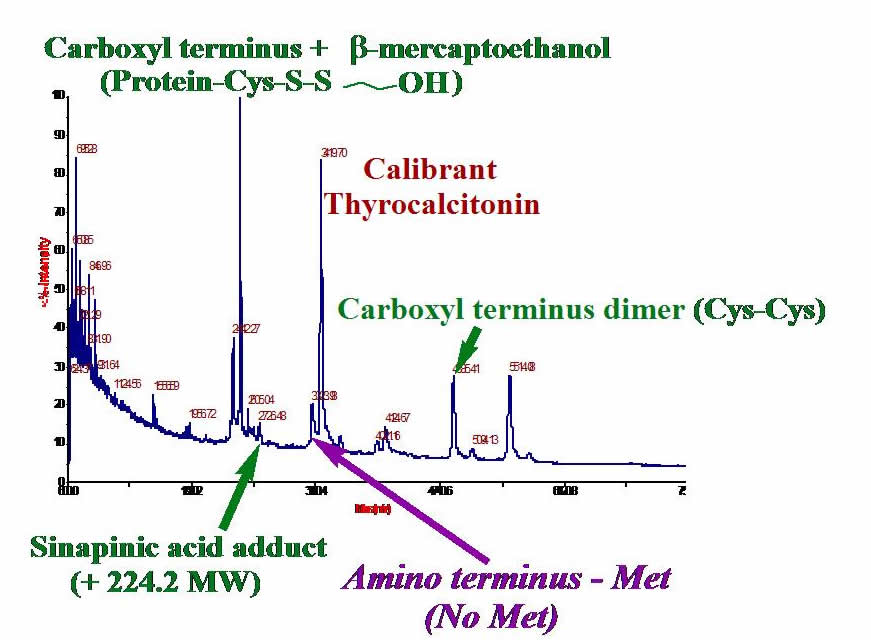
Mass Spectrometry. Measuring the masses of your proteins can give you valuable information about whether or not you have the expected protein. Often, post-translational modifications occur on a protein, so sequencing proteins using MS is very useful. We have developed techniques for using MALDI-TOF mass spectrometry to gain masses of extremely hydrophobic proteins. The graphic shows MALDI-TOF of one of our proteins: the cystine dimer, matrix adduct, mercaptoethanol adduct, and protein proteolyzed at the methionine amino terminus are detected. We also use electrospray ionization MS for studying protein fragments.
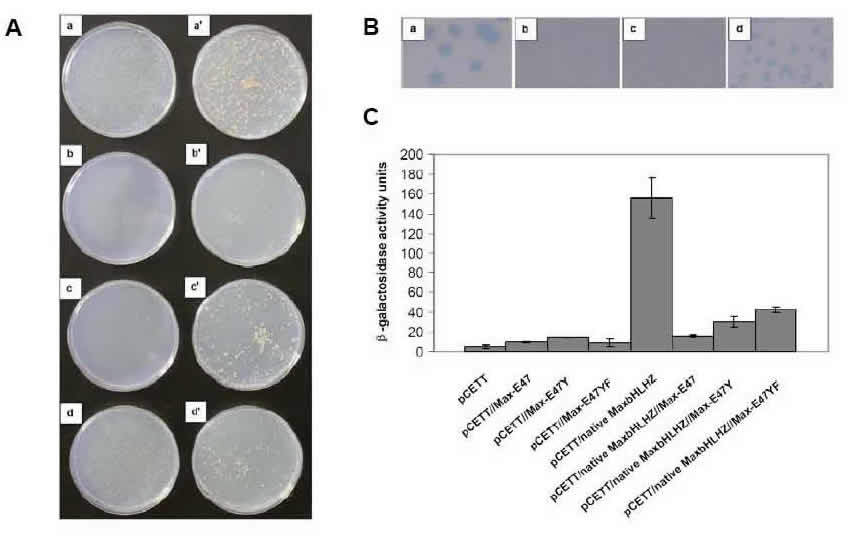
Yeast Reporter Assays. We use both the yeast one-hybrid and two-hybrid assays (as well as our modified yeast reporter system) to assay for protein:DNA and protein/protein interactions, respectively. Thus, we use the above in vitro techniques to detect and quantify protein interactions with DNA, and in vivo yeast assays to measure activity in live cells. We perform a number of different yeast measurements: first, we use the HIS3 selection that assays for cell survival on plates (the graphic shows the plates on the left side). Then we use the sensitive X-gal colony-lift assay, in which the intensity of blue color indicates strength of protein:DNA binding (colony-lift shown at top right of graphic). We perform the quantitative ONPG assay to gain actual numbers correlating to protein:DNA binding affinities in live yeast cells (data displayed as histogram). All three of these yeast assays (and others) are performed to assay activity in the physiologically relevant cellular environment.
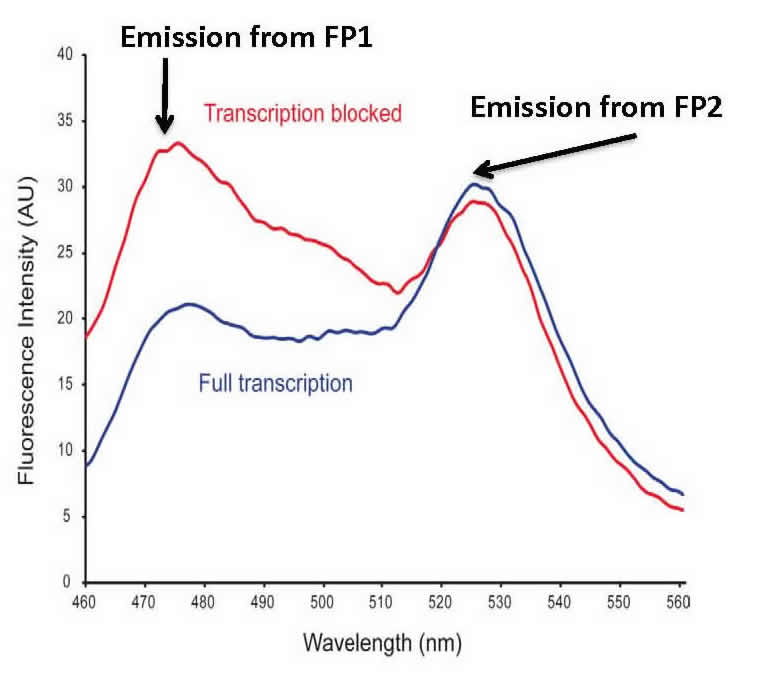
Fluorescence Resonance Energy Transfer (FRET). We also test protein:DNA and protein/protein binding in E. coli cells. Our bacterial cell assay uses two fluorescent proteins that can engage in FRET. Our construct works such that in the presence of bound protein, FRET is decreased or abolished. The graphic shows an overlay of the divergent FRET spectra resulting when transcription is blocked by a bound protein (blue) or when transcription is unaffected (red). Our bioprobe system has the capability of high-throughput library screening of protein:DNA and protein/protein interactions in a straightforward bacterial system.
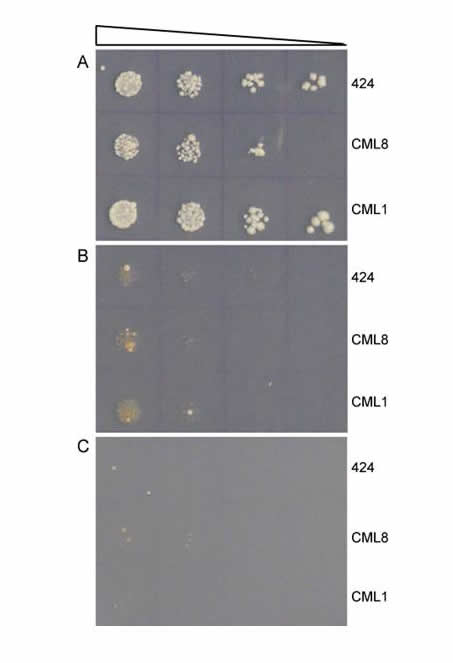
Directed Molecular Evolution. We use rational design to generate new proteins; this process involves extensive structural knowledge about a protein and what features it uses to interact with a ligand. If this information does not exist, rational design is difficult or even impossible. We can combine our rational design capability with directed evolution, in which we allow a diverse library of proteins to compete—e.g. we want to find the protein that binds most efficiently to a desired DNA target sequence. In this "survival of the fittest" scenario, the protein library is expressed in yeast, and ability to bind the DNA target confers the ability for a yeast cell to survive. After extended competition in yeast, we assay isolate and amplify the survivors. The graphic shows a selection where uncovered protein CML8 shows weak growth ability, only slightly higher than the 424 control or another protein CML1. Successive rounds of directed evolution can be performed to select and evolve new and improved protein variants.